血脂异常和由此产生的脂毒性是代谢综合征和2型的病理特征。过多的脂质导致细胞功能障碍,并通过与氧化应激相关的多效性机制诱导细胞死亡。然而,调节新陈代谢应激反应的途径还不是很清楚。
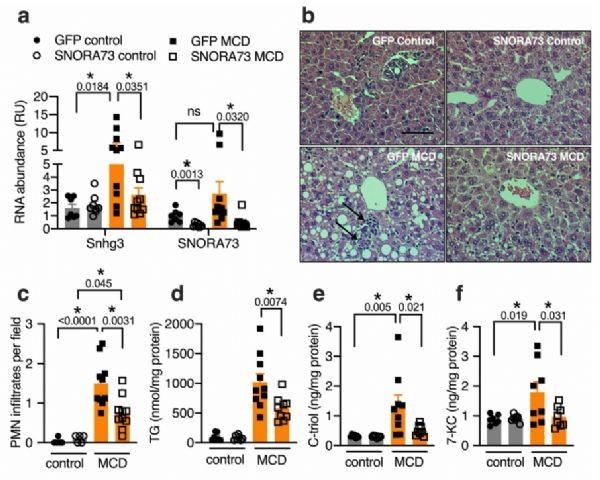
在这里,作者证明了编码在小核仁RNA宿主基因3(Snhg3)内的H/ACA SNORA73盒小核仁RNA的破坏导致了培养细胞对脂质诱导的细胞死亡和一般氧化应激的抵抗。这种对代谢应激的保护与氧化代谢的广泛重新编程有关,而氧化代谢依赖于哺乳动物雷帕霉素信号轴的靶点。此外,作者发现在体内敲除SNORA73可以预防肝脏脂肪变性和脂质诱导的氧化应激和炎症。
在营养过剩的状态下,脂质过载会超过脂肪组织储存甘油三酯的能力和非脂肪组织代谢脂肪酸的能力。这导致异位脂质储存在诸如肝脏,心脏和骨骼肌的组织中。尽管非脂肪细胞中的甘油三酯储存最初起到细胞保护作用,但异位脂肪变性最终与细胞功能障碍和细胞死亡相关,其通过脂毒性过程损害器官功能。对动物模型的研究提供了令人信服的证据,证明脂毒性在非酒精性脂肪性肝病(NAFLD)的发病机制中起作用,NAFLD是2型和代谢综合征最常见的并发症。
脂毒性的特征是应激反应途径被激活,这是由于过度提供底物给脂质利用的生理途径造成的。饱和脂肪酸为神经酰胺的从头合成途径提供燃料,神经酰胺可以启动导致细胞死亡的信号传递。过多的脂质摄取,特别是饱和脂肪酸,会导致内质网(ER)膜的快速重塑,从而损害细胞器的完整性,并导致内质网应激反应的激活。线粒体功能障碍,由不利的膜重塑和增强的底物代谢引起,损害能量产生并启动线粒体的凋亡程序。
内质网应激和线粒体功能障碍都会导致活性氧(ROS)的产生,从而压倒内源性抗氧化机制。饱和脂肪酸诱导的NADPH氧化酶激活,NF-κB介导的促炎细胞因子转录和死亡受体信号传导进一步加剧了ROS的积累。抗氧化剂减轻脂毒性的观察结果支持这样的观点,即氧化应激是促进脂毒细胞死亡的关键因素。从系统上看,内质网和氧化应激信号通路与炎症信号通路的交叉也会导致慢性低度炎症。尽管如此,控制脂毒反应的近端分子转导仍然没有完全确定。
基因筛选确定了SNORA73在调节细胞对脂毒性和氧化应激反应中的作用。作者使用的启动子陷阱突变策略能够破坏标准全基因组shRNA或CRISPR筛查中通常不针对的非编码元件,可能是因为snoRNA宿主位点倾向于作为前病毒和其他可移动元件的整合位点。
体内外研究表明,SNORA73的缺失导致代谢重排和消除脂毒性,揭示了snoRNAs调节细胞代谢和代谢应激的途径。作者的发现提出了这样一种可能性,即snoRNA丰度或功能的变化可能是人类代谢表型变异的基础,这些非编码RNA可能成为的治疗靶点。(来源: Bioon.com)